
 购物车
购物车


- 首页
- >
- GENESEED
- >
Nature丨“猎环人”如何应对circRNA研究的各种挑战?
2024年11月11日,美国加州洛杉矶自由科学记者Amber Dance在Nature的TECHNOLOGY FEATURE专栏上发表文章:Circular logic: understanding RNA’s strangest form yet,阐述了科学家们在circRNA研究中面临的各种挑战。

circRNA曾被认为是剪接错误的产物,如今人们知道它在生物体中广泛存在,与癌症、心血管疾病和阿尔茨海默病等疾病有关,并为治疗药物和生物标志物提供了广泛的可能性,circRNA引起了广大研究者的兴趣。
科学家们仍在研究circRNA的作用:如清除miRNA,以防止miRNA与mRNA结合并抑制蛋白质的产生;与各种蛋白质或RNA聚合酶相互作用,以调节转录和蛋白质表达;甚至可被翻译(图1)。
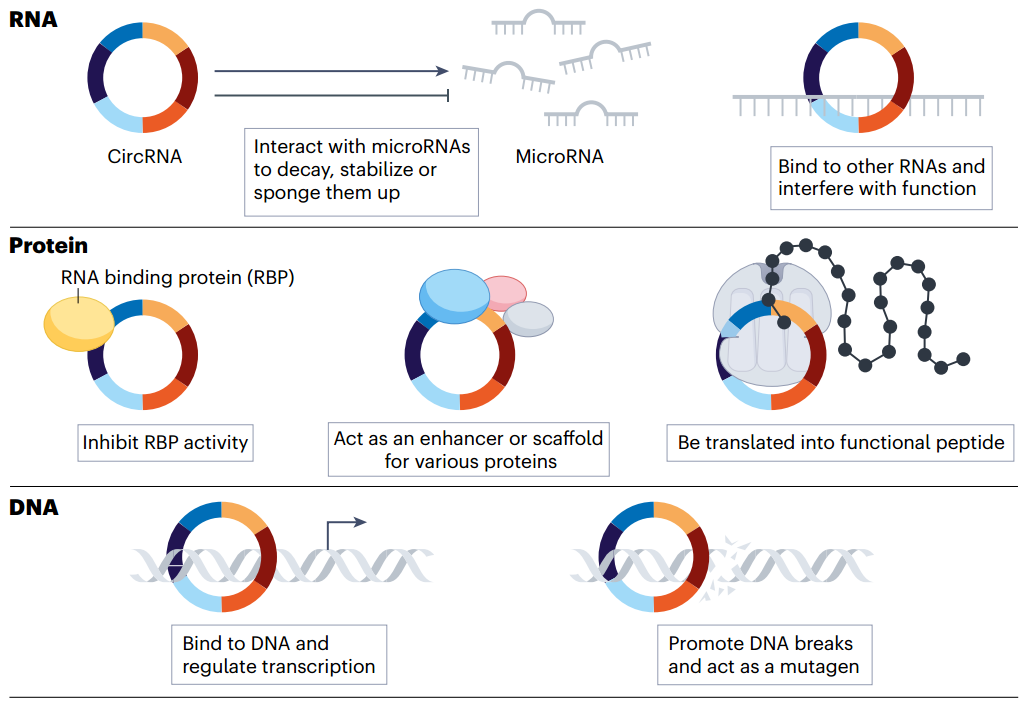
图1 环状RNA在细胞中有多种潜在功能:靶向RNA、蛋白质或DNA分子。
柏林系统生物学家Nikolaus Rajewsky:“关于circRNA,确实有很多有趣的事情有待发现。这是一个未知而又令人感兴趣的领域。”
circRNA的研究极具挑战。circRNA较罕见,约占非核糖体RNA的0.1%[1]。circRNA与相同基因转录的线性RNA的唯一区别在于连接成环处(环化位点)。因此,在线性RNA干扰下,难以分析或生成较纯的环状结构。
刚进入circRNA领域的研究人员可参考已发表的最佳研究。美国耶鲁大学Grace Chen建议,最好方法是着手circRNA研究前咨询circRNA领域的“老手”。
Chen:“我认为,如果人们对这个领域感兴趣,就应该努力进入这个领域。这里有很多待发现的东西。”
circRNA数据库
挑战:circRNA数据库的选择
比利时根特大学的癌症基因组学研究员Jo Vandesompele团队在2020年审查了circRNA数据库,他警告:有很多数据库都是未经整理的、不完整的,且充斥着未经验证的circRNA;同一个分子有很多名称,数据库概况完全是个噩梦。
因而选择数据库时需留心,Vandesompele会选择来自中国科学院北京生命科学研究院赵方庆团队的circAtlas,因为circAtlas通过两种工具来识别任何列出的circRNA。
circRNA测序
挑战:circRNA测序方法的选择
测序是寻找新环状RNA的最常用方法,但circRNA不存在于靶向poly(A)尾构建的标准RNA文库中,因为circRNA缺乏这种特征。美国贝勒医学院的RNA生物学家Jeremy Wilusz说:“你需要专门寻找它们。”
最常见的选择是创建总RNA文库,这比构建circRNA特异性文库更容易,成本更低,且可以检测多种RNA形式,不仅是环状RNA。
另一种方法是去除大部分非环状RNA。需要用RNase R,它可以攻击线性RNA的末端。但Vandesompele警告,注意滴定反应条件很重要,酶太多会破坏circRNA,太少则会残留一些线性片段。此外,某些RNA结构,如G-四联体、组蛋白mRNA和小核RNA,也会阻碍RNase R作用。Wilusz团队报告,将反应缓冲液中的钾换成锂可破坏这些RNA结构,并有助于改善线性RNA的降解[2]。
circRNA测序数据
挑战:分析工具的选择
Vandesompele称,美国Illumina开发的短读测序结果一般都比较好。RNA测序之后,就需要在这些序列中寻找关键的环化位点。但各种算法在灵敏度上差别很大。
Vandesompele团队在2023年进行的16种工具的比较,单个算法从同一细胞系中识别出1372到58032个circRNA[3]。Vandesompele建议研究人员至少选择两种假阳性率低的工具。
挑战:测序结果拼接
这些算法都不是完美的, Rajewsky指出:“根据我们的经验,得到的数据中有80%是真实的。”意料之外的拼接形式或放大事件可能会产生虚假的结果。
Vandesompele说,长读测序虽然成本较高,却是另一种很好的选择,只有这样,你才能更精准地识别circRNA。
吉赛生物Nanopore circRNA全长测序服务优惠活动火热进行中,可更精准地识别circRNA——性价比高的明智选择!!
挑战:参考基因组比对
美国斯坦福大学的计算生物学家Julia Salzman说,另一个问题是,大多数搜索算法依赖于将RNA序列与参考基因组比对。如果参考基因组中缺少circRNA序列,可能会产生假阴性结果。如果基因组中可能有接近同源的序列,试图匹配这些序列就可能产生假阳性。
Salzman团队开发了一种名为SPLASH2的替代方法。该软件基于被称为k-mers的短基因片段的计数,可在不依赖参考基因组的情况下比较两组序列。
circRNA的结构验证
量化已知circRNA,可使用微阵列芯片,利用核酸探针检测环化位点。Arraystar公司的科学家Yanggu Shi表示,大约70%的微阵列预测是正确的。
通常使用qPCR进一步验证circRNA的存在和环状结构。用RNase R处理样品以降解线性RNA,未经处理的样品作为对照,并扩增两个样品的RNA。真正的circRNA经得起RNase R的处理,而线性的对应物则会被酶切。
挑战:多种类型的circRNA结构检测
环化位点的检测只代表一部分的circRNA,因为可能存在多个环有不同的外显子,甚至内含子共享相同的环化位点。
因而Wilusz的验证策略包括非PCR方法,如Northern blotting,分离不同大小的RNA分子,然后使用序列特异性探针检测感兴趣的分子。这样不仅可检测特定的RNA,还可确定有多少不同大小的分子含有这些序列。
circRNA的丰度验证
澳大利亚弗林德斯大学分子生物学家Vanessa Conn说,circRNA的丰度可为其功能提供重要线索。例如,如果circRNA可清除miRNA,那么circRNA应该足够丰富,才能捕获细胞中的大多数miRNA。但即使是一种罕见的circRNA也可能影响基因的表达。
挑战:不同大小的circRNA的量化
弗林德斯大学的研究人员说,量化circRNA可能很棘手,因为样本中的小分子比大分子的circRNA扩增更快,使小分子显得更丰富。
为解决这个问题,Conn团队开发一种名为SplintQuant的方法[4]。通过设计与目标circRNA接头配对的DNA探针,然后用一种酶将发现circRNA的探针对连接在一起,并利用qPCR进行计数。
挑战:不同研究的结果对比
但是如何对比不同实验室和操作流程的circRNA序列和量?
Conn团队合成称为SynCRS的circRNA的形式,将已知量的SynCRS加入到建库前的样品中[5],就可将不同实验室的结果标准化。
circRNA的功能研究
挑战:circRNA去除和过表达方式
关键的方法包括敲除或过表达circRNA。难度还是在于与circRNA相似其他RNA类型,会把池子搞混,混淆视听。
① circRNA干扰
墨西哥Federico Gómez儿童医院的医学研究员Guillermo Aquino-Jarquin说,RNA干扰技术可靶向环化位点,是常用的技术。也可设计干扰RNA,特异性地结合和抑制怀疑的核酸或蛋白质的结合位点。
另一个选择是使用CRISPR-Cas系统,靶向敲除目标circRNA的基因。但线性RNA的产生也会受到基因组改变的影响。
研究人员也可利用Cas13酶靶向RNA水平上的环化位点或其他结合位点。Aquino-Jarquin说:“这样敲掉circRNA,但没有破坏基因组”。他估计,这种敲除方式的有效性约为80-90%,而RNA干扰的有效性为50-60%。然而,Cas酶也会有脱靶效应。
② circRNA过表达
Wilusz说,对circRNA的验证仍是关键。例如,那些认为circRNA可能吸附miRNA的研究人员,会去除circRNA去寻找结果,这也突变了可能的miRNA结合位点。如果去除一个circRNA会产生一种表型,那么添加这个circRNA应该会逆转。这就是需要过表达的原因。Rajewsky说:“这是可行的,但不可能做到完美。据我所知,无人能生产100%干净的circRNA,因为会引入一些其他RNA。”
Obi称,研究人员可体外合成circRNA,也可设计质粒利用细胞自身剪接机制体内产生circRNA。后者在理想情况下,最真实地再现它在细胞中的产生。另一种经典的方法是排列内含子-外显子(PIE)策略,它包括将外显子和内含子重新排列促进circRNA产生。
Obi说,为了更好地控制circRNA的纯度,实验室的体外合成方法更可取。Obi和Chen更倾向于生成线性RNA前体,然后使用连接酶将其连接。为尽量减少副作用,研究人员可添加RNase R以降解不需要的线性RNA,并使用凝胶电泳或液相色谱等分离技术进行纯化。
③ 验证技术
挑战:功能验证方法是否合适
但Rajewsky警告,合成circRNA,放入的任何东西都是人为的,可能与生命活动完全无关。

因此,关键是用多种技术进行验证。例如,Rajewsky团队最近报道circRNA Cdr1as与miR-7相互作用,调节神经元中神经递质谷氨酸的释放[6],其中应用了多种技术:哺乳动物初级神经元培养、化学和电刺激、单细胞RNA成像和miRNA敲除——仅举几例。
Rajewsky:“这是不是有点太过了?对circRNA而言一点都不为过,有必要对这些crazy的分子说些实在的!”
原文链接
https://www.nature.com/articles/d41586-024-03683-w
参考文献
[1] Gruner, H., Cortés-López, M., Cooper, D. A., Bauer, M. & Miura, P. Sci. Rep. 6, 38907 (2016).
[2] Xiao, M. S. & Wilusz, J. E. Nucleic Acids Res. 47, 8755-8769 (2019).
[3] Vromman, M. et al. Nature Methods 20, 1159-1169 (2023).
[4] Conn, V. & Conn, S. J. RNA 25, 1202-1210 (2019).
[5] Conn, V. M., Liu, R., Gabryelska, M. & Conn, S. J. Nature Protoc. (2024)
[6] Cerda-Jara, C. A. et al. EMBO Rep. 25, 3008-3039 (2024).

 广州市黄埔区开源大道11号科技企业加速器A区6栋2楼
广州市黄埔区开源大道11号科技企业加速器A区6栋2楼


 geneseed@geneseed.com.cn
geneseed@geneseed.com.cn