
 购物车
购物车


- 空间组
-

- 高通量药筛
-

- 蛋白组
-
- 代谢组
-
- 定位分析
-
- 病毒包装
-
- ·
ATAC-seq
- 产品介绍
- 案例解析
- 结果展示
- 送样建议
服务介绍
通过转座酶对某种特定时空下开放的核染色质区域进行切割,获得在该时空下基因组中所有活跃转录的调控序列,再通过聚类分析,挖掘这些开放位点的潜在结合转录因子,结合基因表达水平数据,发现关键的调控转录因子。
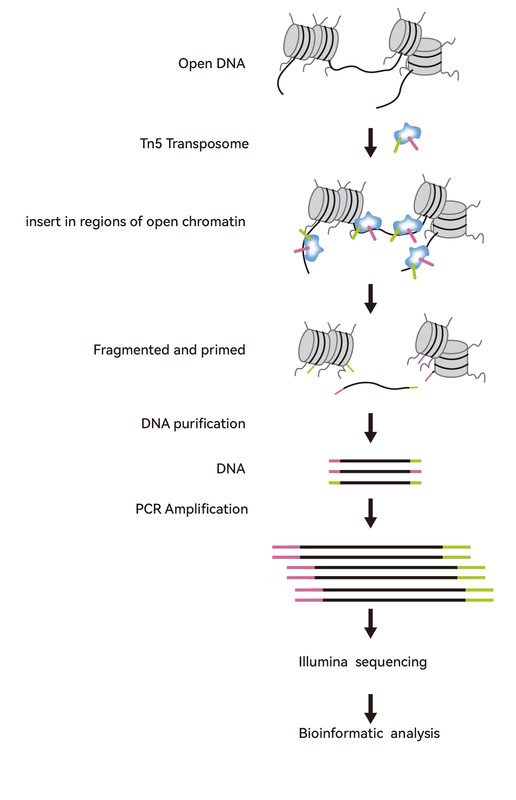
染色质开放性测序技术路线
测序方案
测序平台:Illumina Novaseq 6000/NovaSeq X Plus测序模式:PE150
测序数据量:12~15 Gb raw data
ATAC-seq + RNA-seq 关联分析
DNA调控元件活性的一个特征就是染色质可接近性,只有活跃的调控元件才能被细胞机制识别表达,因此,染色质的可接近性与转录调控密切相关。ATAC-seq 可以得到某一时空下全基因组染色质开放区域,RNA-seq可以得到同一时空下基因表达信息,通过联合两个组学的数据,可以获得该时空下影响基因表达的的上游调控区域;并且调控元件富集数据与基因表达数据能相互验证。

2016年6月,清华大学的颉伟老师领导的团队在国际顶级学术期刊《Nature》发表了染色质可接近性研究(IF:40.137),可以说是用ATAC-seq寻找关键转录因子的经典教材。该文章联合ATAC-seq和RNA-seq检测了早期胚胎发育过程中各时期染色质结构开放区域的动态变化,发现不同时期的细胞开放区域的动态变化情况,并得到胚胎发育时期关键转录因子,绘制出了小鼠早期胚胎中全基因组易接近性染色质图谱。
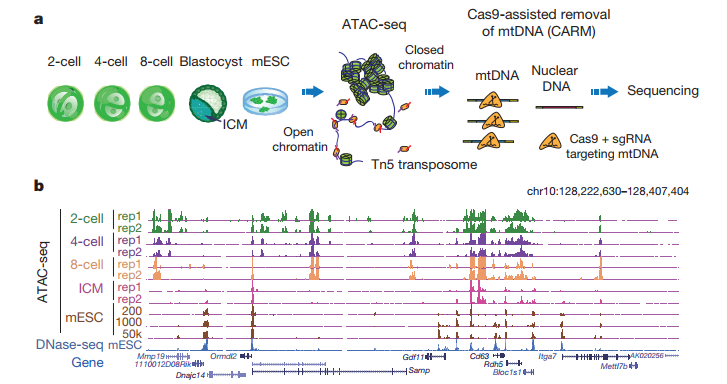
Nature, 2016, 534(7609): 652-7.
Figure1.小鼠着床前胚胎细胞染色质开放性可视图

Nature, 2016, 534(7609): 652-7.
Figure2.胚胎发育过程各时期关键调控元件及转录因子预测结果
ATAC-seq技术是一种快速和灵敏的表观基因组学研究方法,只需要少量细胞即可捕获全基因组活性调控序列。因此,对于临床上珍贵而稀少的细胞样本(如胚胎细胞)的染色质状态的研究就显得非常有利而重要,并且能够很好的推进染色质可接近性研究在基础研究与临床开发中的应用。
生物信息分析
基础分析
原始序列数据
测序数据质量评估过滤
测序数据比对到参考基因组
结合位点peak注释分析
motif注释分析
结合位点peak关联基因筛选
含分组样本时可添加这些分析
组间数据差异peak分析
差异结合位点peak识别
差异结合位点peak关联基因鉴定
差异位点peak靶基因的 GO 功能富集分析
差异位点peak靶基因的 KEGG 富集分析
差异位点peak靶基因的 Reactome 富集分析
高级分析
转录因子motif足迹分析
部分结果示例
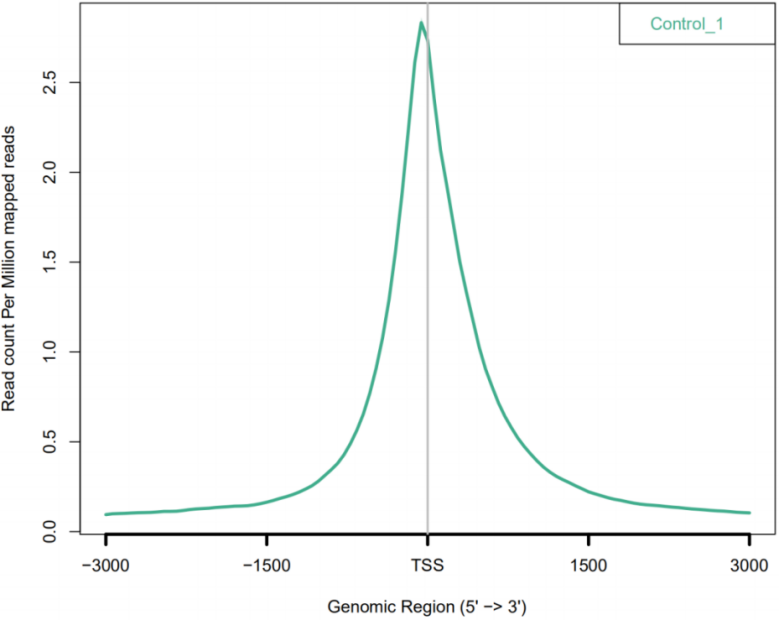
图1. reads相对TSS位置的分布

图2. 插入片段长度展示
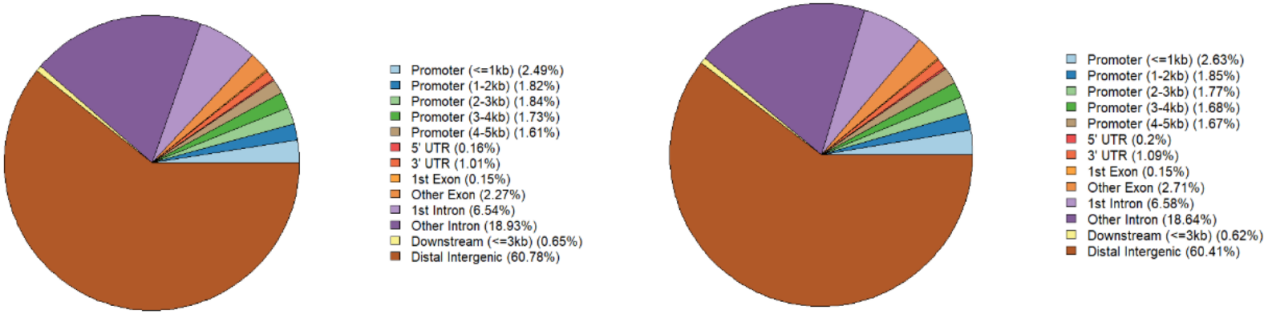
图3. peak 在功能区域上的分布展示
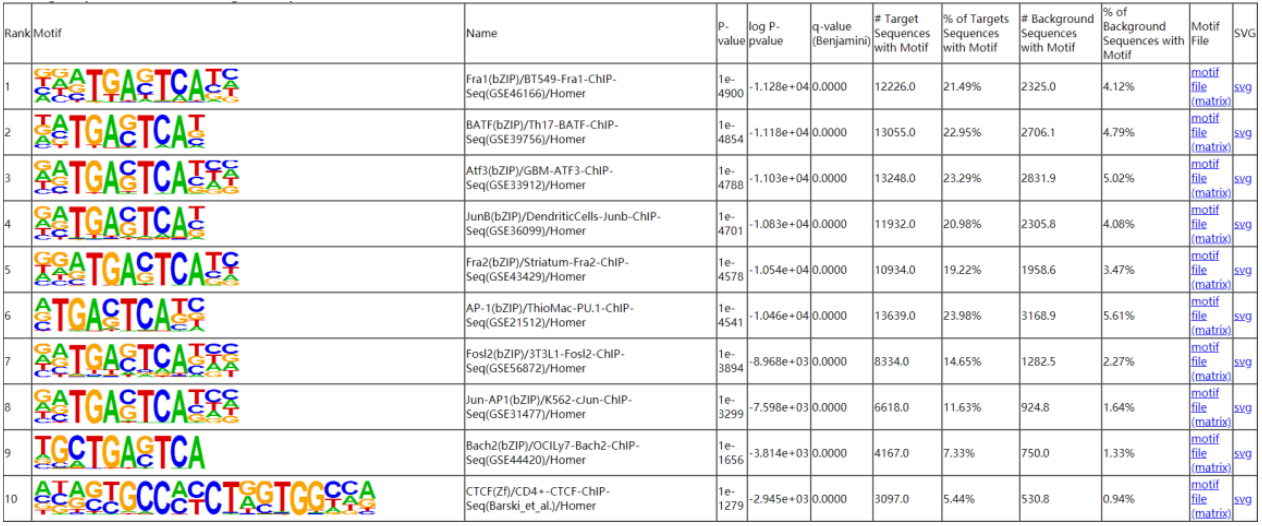
图4. Motif分析结果展示
Table1.ATAC-seq原始样本送样建议
送样类型
送样量
备注
细胞 (细胞数)
≥2*10^6;活性大于80%
无支原体污染
动物组织
≥200mg
植物组织 ≥200mg *更具体的送样方法请详询销售或技术支持
物种范围:人、小鼠、大鼠,其他物种请咨询销售或技术支持
填写需求描述给我们
工具快速咨询
400-8989-400
geneseed@geneseed.com.cn

 广州市黄埔区开源大道11号科技企业加速器A区6栋2楼
广州市黄埔区开源大道11号科技企业加速器A区6栋2楼


 geneseed@geneseed.com.cn
geneseed@geneseed.com.cn